

In the world of medical technology, the path to market begins with one critical question: How does the FDA classify your device? Under the Electronic Code of Federal Regulations (eCFR), specifically Title 21, Part 860, the FDA outlines the formal procedures used to determine the regulatory "class" of a medical device.
At Satori One Click Solutions LLP, we help businesses navigate these complex legal frameworks with ease. This article provides a detailed breakdown of Part 860 to help you understand the classification process and ensure your product meets federal standards.
What is 21 CFR Part 860?
Part 860, titled "Medical Device Classification Procedures," is the regulatory roadmap the FDA uses to categorize devices based on the level of control necessary to assure their safety and effectiveness. This classification is vital because it determines whether your device requires a 510(k) premarket notification, a Premarket Approval (PMA), or if it is exempt from certain requirements.
The regulation is organized into four key subparts:
- Subpart A – General: Sets the foundational rules and definitions for classification.
- Subpart B – Classification: Details the specific procedures for assigning a device to Class I, II, or III.
- Subpart C – Reclassification: Explains how a device's classification can be changed as new safety data emerges.
- Subpart D – De Novo Classification: Provides a pathway for novel devices that do not have a "predicate" (a similar device already on the market).
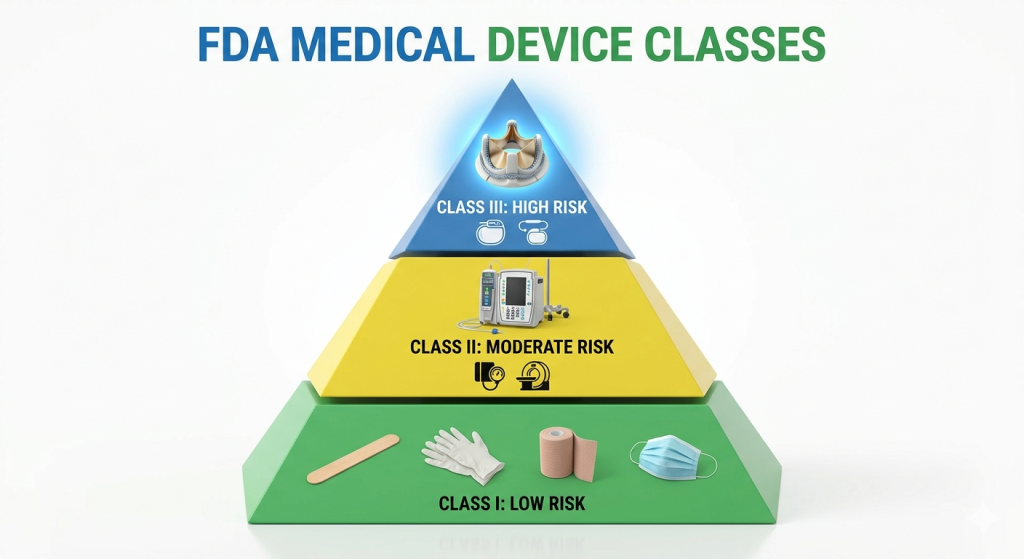
The Three Tiers of Medical Device Classification
While Part 860 focuses on the procedures for classification, it is built around the three-class system used by the FDA:
- Class I (General Controls): These are low-risk devices, such as bandages or handheld surgical instruments.
- Class II (Special Controls): These carry moderate risk and often require a 510(k) submission to prove they are "substantially equivalent" to an existing device. Examples include infusion pumps or powered wheelchairs.
- Class III (Premarket Approval): These are high-risk or life-sustaining devices, such as replacement heart valves or implanted cerebelar stimulators, which require the most stringent level of FDA oversight.
Modern Paths: De Novo and Reclassification
One of the most important sections of Part 860 for innovators is Subpart D: De Novo Classification. If you have a low-to-moderate risk device but there is no similar product already cleared by the FDA, the De Novo pathway allows you to request a new classification rather than being automatically placed into Class III.
Additionally, Subpart C provides the mechanism for reclassification. As the medical community gains more Real-World Evidence, the FDA may move a device from Class III to Class II if it is determined that "Special Controls" are sufficient to ensure safety.
Why Your Business Needs a Classification Strategy
Choosing the wrong classification path can lead to months of delays and thousands of dollars in wasted resources. To be successful, your digital and regulatory strategy should follow Search Essentials:
- Organize Your Documentation: Group similar technical data topically to help regulators (and search engines) understand your product's purpose.
- Use Clear Language: Write your indications for use naturally, avoiding "keyword stuffing" or overly complex jargon that might confuse your filing.
- Corroborate with Expert Sources: Always link your internal data to official resources like the eCFR or FDA Guidance Documents to build trust and reliability.
Streamline Your Success with Satori One Click Solutions LLP
Navigating 21 CFR Part 860 doesn't have to be a burden. Satori One Click Solutions LLP offers specialized B2B services to help you identify the correct classification for your medical device, prepare your Subpart B or Subpart D filings, and maintain compliance throughout the product lifecycle.
We provide a "one-click" experience for regulatory hurdles, ensuring your life-saving innovations reach the people who need them most. Contact Satori One Click Solutions LLP today to start your journey toward FDA clearance with confidence.
Get in touch with us today and take the first step toward FDA clearance:
📧 Email: satoriocs@gmail.com
🌐 Website: https://satoriocs.com
📞 Phone: +91-9829098077 / 9216598077