 +91 98290 98077
+91 98290 98077 +1-519-498-5957
+1-519-498-5957 +1-603-203-0935
+1-603-203-0935Medical Devices Regulations and Licensing Global
Welcome to Satori OCS LLP, your trusted partner in navigating the complex and ever-evolving landscape of global medical devices regulatory and licensing. In an era where healthcare technology is advancing at an unprecedented pace, ensuring that your medical device meets regulatory requirements and receives the necessary approvals is critical to market success and, most importantly, patient safety. With a deep understanding of international regulations, a team of seasoned experts, and a commitment to excellence, we stand ready to guide you through the intricate maze of compliance, helping you bring life-saving innovations to the global market with confidence and compliance. Discover how Satori OCS can be your strategic ally in this dynamic and highly regulated industry.

What are Medical Devices ?
Medical devices are instruments, apparatus, machines, equipments or other products that are used to diagnose, monitor, prevent, treat or alleviate medical conditions or diseases. They can be simple or complex and range from items/ simple tools/ surgical instruments, such as tongue depressors, stethoscopes and bandages and assistive devices such as hearing aids and wheelchairs to high-tech devices/ complex machines/ diagnostic equipments such as magnetic resonance imaging (MRI) machines, X-ray machines, implantable devices such as pacemakers, artificial hearts. The use of medical devices has revolutionized healthcare and has improved the diagnosis, treatment, and management of medical conditions for millions of people around the world. They can be designed to be disposable or reusable, and can be powered by electricity, batteries, or manually. Medical devices can be used in a variety of healthcare settings, including hospitals, clinics, laboratories, and homes. They play a crucial role in healthcare, providing healthcare professionals with the tools they need to diagnose medical conditions, such as blood glucose monitors used to test blood sugar levels, or to treat or manage medical conditions, such as insulin pumps used to deliver insulin to people with diabetes. They are also used to monitor patients and to provide ongoing care for chronic conditions.
Categories of Medical Devices
Medical devices can be categorized into different classes based on the level of risk associated with their use. The classification of medical devices varies depending on the country or region where the device will be sold.
However, some common categories of medical devices include:
- Class I Medical Devices
These are low-risk medical devices that do not pose significant risk to patients or users and do not require regulatory approval before they can be sold. Examples include tongue depressors, bandages, and stethoscopes. - Class II Medical Devices
These are medium-risk medical devices that pose a moderate risk to patients or users and require regulatory clearance before they can be sold. Examples include syringes, surgical gloves, blood glucose meters, contact lenses, X-ray machines and surgical instruments - Class III Medical Devices
These are high-risk medical devices that pose a significant risk to patients or users and require regulatory approval before they can be sold. Examples include pacemakers, heart valves, implantable defibrillators/ devices, and dialysis machines. - Class IV Medical Devices
These are the highest-risk medical devices that pose a significant risk to patients or users and require rigorous regulatory approval before they can be sold. Examples include cardiac stents, artificial heart valves, novel implants, and life-supporting devices/ machines.
Additionally, some countries or regions have subcategories within these main classes, such as Class IIa and IIb in Europe, or Class III and Class IIIa in Canada.
In addition to the above categories, some regulatory authorities and markets may have additional classification systems. For example, the US FDA also has a separate classification system for in vitro diagnostic medical devices (IVDs) based on the level of risk they pose to patients and users.
Medical devices can also be categorized based on their intended use, such as diagnostic devices, therapeutic devices, monitoring devices, surgical instruments, and implantable devices. These categories help to further classify medical devices based on their specific functions and applications.
It is important to note that the classification of a medical device can impact the regulatory requirements for the device, including the level of testing, documentation, and quality control needed to demonstrate safety and efficacy.
Notified and Non-notified Medical Devices under MDR 2017
Medical devices under the Medical Device Regulation (MDR) of 2017 are categorized into two main groups:
- Notified Medical Devices: "Notified" medical devices are those that require the involvement of a Notified Body for conformity assessment. These devices have a higher risk classification and undergo a more comprehensive evaluation to ensure compliance with the MDR's safety and performance requirements. Notified Bodies are independent organizations designated by EU member states to assess the compliance of medical devices with the MDR's requirements. Examples of Notified Medical Devices include implantable devices, Class III devices, active implantable devices, and certain Class IIb devices.
- Non-notified Medical Devices: "Non-notified" medical devices are those that fall under lower risk categories and do not necessitate the intervention of a Notified Body for conformity assessment. These devices still need to meet the essential requirements of the MDR, but the assessment process is generally less extensive compared to "Notified" devices. Manufacturers can self-declare the conformity of these devices with the MDR's requirements and place them on the market. Non-notified devices are typically lower-risk Class I devices, as well as certain Class IIa devices that are not considered high-risk.

Global Laws and Regulations Controlling Medical Devices
Several global laws and regulations govern the manufacturing, distribution, and use of medical devices to ensure their safety, efficacy, quality, and proper functioning.
Here are some of the key international frameworks and regulations controlling medical devices:
-
 European Union (EU) Medical Device Regulation (MDR) and In-Vitro
Diagnostic Regulation (IVDR)
European Union (EU) Medical Device Regulation (MDR) and In-Vitro
Diagnostic Regulation (IVDR)
The European Union's MDR and IVDR are comprehensive regulatory frameworks that establish rules for medical devices and in-vitro diagnostic devices within the EU. These regulations set out requirements for device classification, conformity assessment, quality management, clinical evaluations, post-market surveillance, labeling, and more.
-
United States Food and Drug Administration (FDA) Regulations
The FDA regulates medical devices in the United States under the Federal Food, Drug, and Cosmetic Act (FD&C Act). The agency classifies medical devices into different classes (Class I, II, or III) based on risk, with varying levels of regulatory scrutiny and requirements and ensures their safety and effectiveness through pre-market clearance or approval processes.
GET IN TOUCH
-
Canadian Medical Device Regulations
Health Canada regulates medical devices in Canada through its Medical Devices Regulations. These regulations cover processes for device approvals and registrations, device licensing, establishment licensing, post-market surveillance, and adverse event reporting. The regulations also establish requirements for device safety, effectiveness, and quality.
-
Therapeutic Goods Administration (TGA) Regulations (Australia)
The TGA regulates medical devices in Australia and oversees their market entry, classification, conformity assessment procedures, post-market surveillance, and quality assurance through the Therapeutic Goods Act.
GET IN TOUCH
-
Japanese Pharmaceuticals and Medical Devices Act
The PMDA (Pharmaceuticals and Medical Devices Agency) oversees the regulation of medical devices in Japan under the Japanese Pharmaceutical and Medical Device Act, ensuring their quality and safety. The Act also defines regulatory requirements for the manufacturing, import, and sale of medical devices.
- China's Medical Device Regulations
The NMPA (National Medical Products Administration) regulates medical devices in China and sets out regulatory requirements for device registration, safety evaluations, and quality management. It also oversees device classification, clinical trials, and postmarket surveillance.
- Indian Medical Devices Rules
Manufacture, Import, Sale, and Distribution of Medical Devices in India is governed by the Indian Medical Devices Rules 2017. These rules were introduced by the Central Drugs Standard Control Organization (CDSCO), which is the national regulatory authority for medical devices and pharmaceuticals in India governed under Drugs and Cosmetics Act 1940.
- Ministry of Food and Drug Safety
(MFDS) (South Korea)
The MFDS regulates medical devices in South Korea under the Medical Devices Act, ensuring their safety, efficacy, and quality.
GET IN TOUCH
- Medical Device Authority (MDA) (Malaysia)
The MDA regulates medical devices in Malaysia under the Medical Device Act, overseeing their market entry, post-market surveillance, and quality control.
GET IN TOUCH
- Medical Device Single Audit Program (MDSAP)
MDSAP allows recognized auditing organizations to conduct a single audit of a medical device manufacturer's quality management system, satisfying requirements of regulatory authorities of multiple countries including Australia, Brazil, Canada, Japan, and the United States. It streamlines the auditing process for medical device manufacturers selling in multiple markets.
- World Health Organization (WHO)
Prequalification of In Vitro
Diagnostics
(IVDs)
WHO's prequalification program evaluates the quality, safety, and performance of IVDs, particularly those used in resource-limited settings.
- Global Harmonization Task Force (GHTF) (now IMDRF)
The GHTF was a predecessor to the IMDRF and aimed to harmonize medical device regulations among participating countries. Its documents and principles continue to influence global regulations.
GET IN TOUCH
- International Medical Device Regulators
Forum (IMDRF)
The IMDRF is a global forum (not a legal framework) that facilitates international collaboration and brings together regulatory authorities & stakeholders to harmonize and coordinate medical device regulations. It develops guidance documents and recommendations that help ensure consistency and alignment in the regulation of medical devices among regulatory authority across different countries.
Countries and regions may have their own specific regulations and requirements, and staying informed about these regulations is crucial for manufacturers, distributors, healthcare professionals, and patients.

Medical Device Licensing
Medical device licensing is a mandatory requirement for all manufacturers, distributors, and importers of medical devices in most countries. Medical device licensing is required for ensuring that medical devices meet regulatory requirements and the devices are manufactured and distributed with consistent quality. The requirements and procedures for medical device licensing vary depending on the country or region where you intend to market your product. Here are some general steps for obtaining medical device licensing:
-
Determine Regulatory Requirements/ Identify Applicable Regulations
Determine the regulatory requirements/ identify the applicable regulations and standards for medical device licensing in the country or region where you intend to market your product. This will help you determine the specific licensing requirements and quality management system (QMS) standards that you need to follow. You can consult with a regulatory expert or check the relevant regulatory authorities' websites to determine the requirements. -
Classify Your Medical Device
Determine the classification of your medical device according to the regulatory authority's guidelines. The classification will determine the regulatory requirements and procedures for your device. -
Submit Application
Submit your application for medical device licensing to the regulatory agency or agencies that have jurisdiction over your medical device along with the technical documentation, applicable fees, and any other required information. -
Prepare Technical Documentation
Prepare technical documentation that includes detailed information about your medical device, including its design and manufacturing, product specifications, intended use, testing and validation data, risk assessment documentation and other relevant information. This documentation should meet the requirements of applicable regulations and standards. -
Receive Approvals and Obtain Licenses
Once your application is processed and approved, you will receive a license or certificate that allows you to market your medical device in the country or region. This may include obtaining pre-market approvals, clearances, or certifications.
ISO 13485
ISO 13485 is a standard developed by the International Organization for Standardization (ISO) specifically for the medical device industry. This standard outlines the requirements for a Quality Management System (QMS) that demonstrates an organization's consistent ability to design, manufacture, and distribute medical devices in compliance with customer and regulatory requirements.
Implementing ISO 13485 is crucial for medical device manufacturers, as it not only ensures adherence to global quality standards but also enhances overall operational efficiency and facilitates market access in various countries. However, it's important to note that ISO itself does not directly grant ISO 13485 certification. Instead, certification is awarded by independent certification bodies accredited by ISO or national accreditation bodies. These certification bodies perform audits and assessments of a company's quality management system to determine its alignment with the ISO 13485 requirements. When the quality management system meets these requirements, the certification body issues a certificate of conformity to ISO 13485.

How to Get ISO 13485 ?
Getting ISO 13485 Certification involves implementing a Quality Management System (QMS) that meets the requirements of the standard and then having a third-party certification body assess and verify that the system meets those requirements. It is important to have dedicated resources and support from top management to ensure the successful implementation and maintenance of a QMS that meets the requirements of the standard.
Here are the general steps to follow:
- Familiarize yourself with the ISO 13485 Standard
Review the standard requirements and ensure you understand what is required for a QMS to be compliant. - Develop a Quality Management System (QMS) Develop and implement a QMS that meets the requirements of the ISO 13485 standard. This process includes documenting your processes and procedures, identifying and addressing risks, and monitoring and measuring the effectiveness of your QMS.
- Conduct Gap Analysis through Internal Audits
Conduct internal audits to determine the current state of your QMS and to assess the effectiveness of your QMS and identify any areas that do not meet the requirements of the ISO 13485 standard and that need improvement. - Train Personnel
Train personnel on the requirements of the ISO 13485 standard and how to implement and maintain a QMS that meets the standard. - Corrective Actions
Take corrective actions to address any non-conformities identified during the internal audits. - Select a Certification Body
Choose a certification body that is accredited to assess organizations against the ISO 13485 standard. - Stage 1 Audit
The certification body will conduct a Stage 1 audit to assess the readiness of your organization for certification. This audit includes a review of your QMS documentation and processes. - Stage 2 Audit
The certification body will conduct a Stage 2 audit to assess the implementation and effectiveness of your QMS. This audit includes a review of your QMS in action and interviews with personnel. - Certification
If your organization passes the Stage 2 audit, the certification body will issue a certificate of compliance with the ISO 13485 standard. - Surveillance Audits
The certification body will conduct periodic surveillance audits to ensure that your organization is maintaining compliance with the standard.

Quality Management System (QMS)
A robust Quality Management System (QMS) is a fundamental requirement for medical device registration and regulatory compliance. A well-implemented QMS establishes standardized processes, procedures, and controls to ensure the quality, safety, and efficacy of medical devices. It encompasses various aspects such as design controls, risk management, document management, and post-market surveillance.
Our experts assist in developing and implementing customized QMS tailored to your organization's specific needs, helping you navigate the regulatory landscape with confidence.
Quality Manual Preparation
Quality manual preparation is essential for medical device manufacturers to demonstrate compliance with the relevant regulatory requirements and ensure the quality and safety of their products. It is an important step for ensuring that medical devices meet regulatory requirements and devices are manufactured and distributed with consistent quality.
Here are the general steps for quality manual preparation:
- Determine Quality Management System
(QMS) Requirements
Determine the QMS requirements for medical device manufacturing in the country or region where you intend to market your product. You can consult with a regulatory expert or check the relevant regulatory authorities' websites to determine the requirements.
GET IN TOUCH
- Develop Quality Manual
Develop a comprehensive quality manual that describes your organization's quality management system and outlines your organization's policies and procedures for meeting the QMS requirements, including design and development, production/ manufacturing, testing and validation, document control, corrective and preventive actions, and distribution of medical devices.
- Develop Standard Operating Procedures
(SOPs)
Develop SOPs that describe the specific processes and procedures that are necessary to implement your QMS. These SOPs should be detailed, step-by-step instructions for all aspects of your QMS.
- Implement QMS
Implement the QMS policies and procedures outlined in the quality manual throughout your organization.
GET IN TOUCH
- Monitor and Maintain QMS
Monitor and maintain the QMS to ensure ongoing compliance with the relevant regulatory requirements and continuous improvement in product quality and safety.
GET IN TOUCH
- Perform Risk Management
Perform a risk management assessment of your medical device to identify potential hazards and risks associated with its use. Use the results of this assessment to develop risk management plans and incorporate risk management into your QMS.
- Establish Post-Market Surveillance
Establish a post-market surveillance plan to monitor your medical device's performance and safety in the field. This plan should include procedures for handling complaints, conducting field corrective actions, and reporting adverse events.
- Conduct Internal Audits
Conduct regular internal audits of the QMS to identify and correct any non-conformities or areas for improvement and to ensure that the QMS is being implemented effectively and is meeting the requirements of applicable regulations and standards.
By following these steps for quality manual preparation, you can establish a robust QMS that ensures that your medical device meets the regulatory requirements and is of consistent quality, safe and effective for use by patients.
MDSAP (Medical Device Single Audit Program)
MDSAP (Medical Device Single Audit Program) is a program that allows medical device manufacturers to undergo a single audit that satisfies the regulatory requirements of multiple countries, including the United States, Canada, Brazil, Japan, and Australia.
The MDSAP offers a streamlined approach for manufacturers seeking to market their medical devices in multiple countries, including Australia, Brazil, Canada, Japan, and the United States. Under MDSAP, a single comprehensive audit is conducted, satisfying the regulatory requirements of participating countries. MDSAP certification is granted by authorized Auditing Organizations (AOs) recognized by the participating regulatory authorities, such as the Therapeutic Goods Administration (TGA) in Australia, Health Canada in Canada, Agência Nacional de Vigilância Sanitária (ANVISA) in Brazil, Ministry of Health, Labor and Welfare (MHLW) and Pharmaceuticals and Medical Devices Agency (PMDA) in Japan, and the United States Food and Drug Administration (US FDA) in the United States.

An authorized auditing organization can conduct an MDSAP audit of a medical device manufacturer, and if the manufacturer passes the audit and meets the program's requirements, they receive an MDSAP certificate. This certificate demonstrates that the manufacturer's quality management system conforms to the requirements of ISO 13485 and the regulatory requirements of the participating MDSAP countries. In essence, MDSAP is a program that allows medical device manufacturers to undergo a single audit that satisfies the regulatory requirements of multiple countries, making it an efficient and effective way to expand market presence.
How to Get MDSAP ?
To obtain the Medical Device Single Audit Program (MDSAP) certification, medical device manufacturers must follow these steps:
The MDSAP program is available to medical device manufacturers who have facilities in one or more of the participating countries, which currently include the United States, Canada, Brazil, Japan, and Australia.
The MDSAP program requires that audits be conducted by an authorized auditing organization. The auditing organization must be recognized by the participating countries. Manufacturers can choose an AO from a list of authorized organizations published by the International Medical Device Regulators Forum (IMDRF).
Prepare for the audit by reviewing the MDSAP audit model and assessing your company's compliance with the applicable regulations and standards, including ISO 13485 and ensuring that their quality management system meets the regulatory requirements of the participating countries.
Once the manufacturer has chosen an AO and prepared for the audit, they can schedule the audit with the AO.
The auditing organization will conduct the MDSAP audit according to the MDSAP audit model, which includes a review of the manufacturer's quality management system and manufacturing processes.. The audit will also assess your company's compliance with the applicable regulations and standards.
After the audit is complete, the AO provides the manufacturer with an audit report that summarizes the findings of the audit.
Correct any non-conformities identified during the audit and provide evidence to the auditing organization that the non-conformities have been corrected.
Once the auditing organization has reviewed the corrective actions and confirmed that the non-conformities have been corrected, they will issue an MDSAP certificate.
The manufacturer submits the audit report to the regulatory authorities in the participating countries to demonstrate compliance with the regulatory requirements.
Maintain MDSAP certification by participating in ongoing surveillance audits and maintaining compliance with the applicable regulations and standards.
The MDSAP certification process can be complex and time-consuming and requires careful planning and preparation. Medical device manufacturers may choose to work with a consultant that has experience with the MDSAP program to help guide them through the process. Our MDSAP auditing services enable you to expand your market presence efficiently and effectively.
How to get Medical Devices Manufacturing Licence in Canada ?
Obtaining a Medical Device Manufacturing License (MDML) in Canada involves a process that is regulated by Health Canada's Medical Devices Bureau.
Following is the general outline of the process:
- Determine the Class of Medical Device
Medical devices in Canada are categorized into different classes (Class I, II, III, and IV) based on their risk level. Each class has different regulatory requirements. Identify the class of your medical device to understand the specific requirements that apply to your situation.
GET IN TOUCH
- Quality Management System (QMS)
Implement a quality management system that complies with the ISO 13485 standard. This system should cover all aspects of manufacturing, including design, development, production, installation, and servicing of medical devices. Health Canada places significant importance on having a well-established QMS.
- Prepare Technical Documentation
Create detailed technical documentation for your medical device. This documentation should cover aspects such as device specifications, manufacturing processes, risk assessment, labeling, and instructions for use.
GET IN TOUCH
- Submit an Application
Prepare and submit an application for a Medical Device Manufacturing License (MDML) to Health Canada's Medical Devices Bureau. The application should include comprehensive information about your company, your medical device, and your quality management system.
- Device Licensing
If your application is approved, Health Canada will issue a Medical Device Establishment License (MDEL) for your company. This license allows you to manufacture, distribute, or import medical devices in Canada. You need an MDEL even if you're the manufacturer. You might also need to list your medical devices in Health Canada's Medical Devices Active License Listing (MDALL) database.
- Regular Audits and Inspections
Health Canada conducts regular audits and inspections to ensure ongoing compliance with regulations and the quality management system.
GET IN TOUCH
- Post-Market Surveillance
After obtaining the MDML, you need to have systems in place for monitoring the performance of your devices on the market and reporting any adverse events to Health Canada.
GET IN TOUCH
- Stay Informed
Health Canada's regulations and requirements can change, so it's crucial to stay informed about updates and changes that might affect your manufacturing license. Regularly review Health Canada's website and updates.
It's recommended to seek guidance from regulatory experts to navigate the process successfully. Our regulatory experts guide you through the MDML process, ensuring that your application meets the Health Canada requirements for getting MDML and facilitating timely market entry of your product in Canada.
510(k) Submissions
Medical device manufacturers intending to market their products in the United States must undergo a 510(k) submission process to the U.S. Food and Drug Administration (FDA). This process is specifically designed for certain medical devices considered low to moderate risk for use in the U.S. The purpose of a 510(k) submission is to demonstrate that the new medical device is substantially equivalent (SE) to an existing legally marketed device, often referred to as a predicate device. The submission is a premarket notification required to ensure that the new device does not pose any unreasonable risks to patient safety or effectiveness. The process for submitting a 510(k) typically involves the following steps:

- Identify a Predicate Device
The manufacturer identifies a legally marketed device that is similar to their device and serves as the predicate device for comparison. The predicate device must have been legally marketed in the U.S. before the submission date of the new device. - Prepare the 510(k) Submission
The manufacturer compiles a comprehensive submission package that includes detailed information about the new device, its intended use, design and performance characteristics, labeling, and any relevant clinical data or testing results. The submission package must demonstrate that the new device is substantially equivalent to the predicate device in terms of safety and effectiveness. - Submit the 510(k) to the FDA
The manufacturer submits the 510(k) package to the FDA electronically or in writing, along with the required user fees. The FDA then reviews the submission to determine if it is complete and whether the device meets the criteria for substantial equivalence. - FDA Review
If the submission is accepted, the FDA reviews the 510(k) package to assess the safety and effectiveness of the new device. This may involve a thorough review of the technical, scientific, and clinical information provided in the submission, as well as any additional information or data requested by the FDA. - FDA Decision
Based on the review, the FDA will issue a decision in the form of a clearance letter or a request for additional information (referred to as a "deficiency" or "hold" letter). If the device is found to be substantially equivalent to the predicate device and meets all other requirements, the FDA will issue a clearance letter, allowing the manufacturer to market the device in the U.S.
It is important for manufacturers to carefully prepare and submit a thorough and complete 510(k) package to help ensure a timely review process. Our regulatory experts guide you through the 510(k)-submission process, ensuring that your application meets the FDA's requirements and facilitating timely market entry in the US.
CE Certificate/CE Mark/ CE Marking
CE stands for "Conformité Européene," which is French for "European Conformity." This term is associated with a certification mark known as a CE certificate, CE mark, or CE marking.
CE certification is a process by which a manufacturer declares that its medical device complies with the essential requirements of the applicable EU directives or regulations, such as the MDD or MDR. It allows the manufacturer to affix the CE marking on their device, indicating that it meets the necessary safety, performance, and quality standards to be sold within the European market.
This mark is essential for certain products, including medical devices, intended to be legally sold within the European Economic Area (EEA). The CE marking signifies that the product complies with European Union (EU) health, safety, and environmental protection standards, meeting the essential requirements outlined in relevant EU directives and regulations.
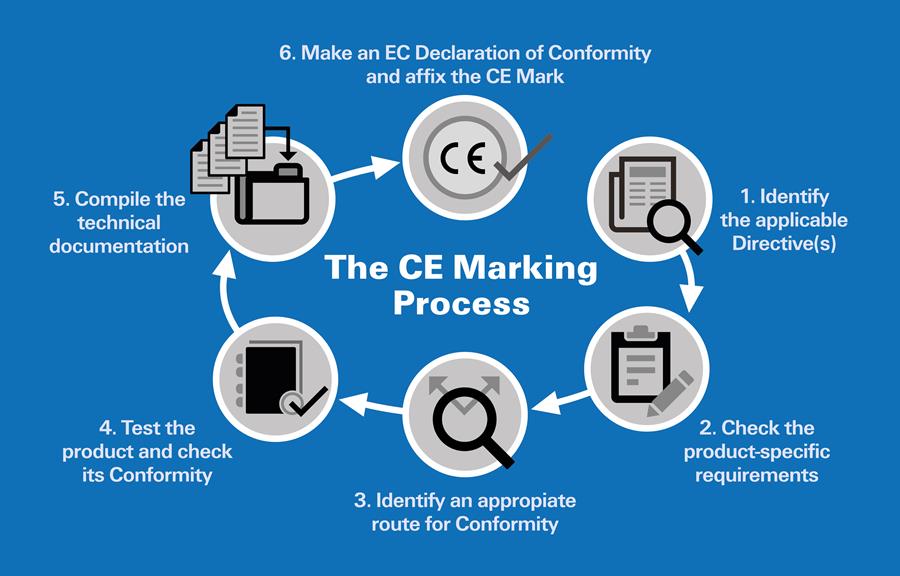
It's important to note that the CE marking is not granted by a regulatory body but is a selfcertification mark affixed by the manufacturer or their authorized representative on the product and packaging. However, the product's conformity with EU directives and standards is assessed by a Notified Body, a third-party organization designated by an EU Member State. The Notified Body issues a certificate or report confirming conformity, and based on this, the manufacturer or their authorized representative issues the Declaration of Conformity and applies the CE marking to the product.
How to Get CE Mark ?
Obtaining CE certification for a medical device involves a series of steps to demonstrate that your device complies with the essential requirements outlined in the European Medical Device Directive (MDD) or the Medical Devices Regulation (MDR), depending on the applicable regulations.
Here is a general outline of the steps to obtain CE certification:
- Determine the Applicable Directives
Identify the specific EU directives and regulations that apply to your product. These directives outline the essential requirements that your product must meet in order to obtain the CE mark.
GET IN TOUCH
- Classification of the Medical Device
Determine the classification of the medical device according to the European Medical Devices Regulation (MDR). Devices are categorized into Classes I, IIa, IIb, or III, with increasing levels of scrutiny and requirements for higher-risk devices. The classification will determine the conformity assessment route and the involvement of a Notified Body.
- Conformity Assessment
Identify the appropriate conformity assessment procedure according to the classification of the device. This involves testing and assessing your product's compliance with the relevant EU directives. The assessment procedures include self-certification by manufacturer themselves (for Class I devices), conformity assessment by a Notified Body (for higher-risk devices belonging to Class Is, Im, IIa, IIb, III), or a combination of both.
- Quality Management System (QMS)
Implement a QMS that complies with the requirements of the MDR. This involves establishing procedures for design control, risk management, supplier management, postmarket surveillance, and more. For certain classes of devices, compliance with ISO 13485 is mandatory.
GET IN TOUCH
- Technical Documentation / File
Compilation for CE Certification
Assemble the technical file or design dossier that includes all the relevant documentation, assessments, and evidence of compliance. Compile all necessary technical documentation that demonstrates your product's compliance with the relevant EU directives and standards. This documentation includes a detailed description of the device, its design specifications, intended use, manufacturing processes, risk assessment/ analysis, and clinical evaluation data (if applicable), labeling, test reports, and instruction for use/ user manuals.
- Clinical Evaluation and issue of Clinical
Evaluation Report (CER)
Conduct a clinical evaluation/ assessment for the higher-risk medical device involving a systematic review of clinical data to determine the device’s safety and performance when used as intended and prepare a CER summarizing the supporting clinical data.
GET IN TOUCH
- Declaration of Conformity (DoC)
The manufacturer must create and issue a DoC, which is a document stating that the medical device fulfils the essential requirements of the MDR and complies with the relevant EU directives and standards. This declaration should include information about the product, its manufacturer, and any applicable standards or specifications.
- Appointing an Authorized Representative
(AR)
If you are a non-EU manufacturer, you must appoint an Authorized Representative located within the EU to act on your behalf.
GET IN TOUCH
- Appointing a Notified Body (if applicable)
For higher-risk devices (Class IIa, IIb, and III), you will need to involve a Notified Body. Choose a Notified Body, and submit the required documentation for review and conformity assessment.
GET IN TOUCH
- Affixing the CE Mark
After obtaining CE certification, affix the CE mark on your medical device, packaging, and associated documentation. The CE mark indicates that your device meets the essential requirements for CE certification and is allowed to be placed on the European market. The mark should be visible, legible, and indelible. It should also be accompanied by other required information, such as the identification number of the notified body involved in the conformity assessment (if applicable).
-
Post-Market Surveillance (PMS) and
Vigilance
Develop and implement a PMS system to monitor the device's performance once it is on the market. This system helps identify and address any potential safety, quality or performance issues and take appropriate corrective actions if any non-conformities are identified.
- Continued Compliance
Ensure ongoing compliance with the MDR, including adherence to post-market surveillance and vigilance requirements.
GET IN TOUCH
- Keep Records
Maintain records of all relevant documentation, including test reports, certificates, and Declarations of Conformity. These records should be kept for a specified period of time (usually 10 years) and made available to authorities upon request.
It is important to note that the requirements and procedures for obtaining a CE mark can vary depending on the product category and the specific EU directives that apply. Seeking the assistance of regulatory experts or consultants can be beneficial to navigate the certification process smoothly. Our regulatory experts guide you through the CE marking process, ensuring that your application meets the EU's requirements and facilitating timely market entry in the EU countries.
At Satori OCS, our mission is to empower your medical device ventures with the knowledge, expertise, and support needed to navigate the intricacies of global regulatory and licensing requirements. We understand that success in the healthcare industry is not just about achieving compliance; it's about enhancing patient care and driving innovation. With a commitment to excellence, a client-centric approach, and a dedication to staying at the forefront of regulatory changes, we are here to be your trusted partner on this journey. As you aspire to make a meaningful impact on the world of healthcare, let us handle the complexities of compliance, so you can focus on what truly matters – advancing medical technology and improving lives. Contact us today to embark on a seamless and successful regulatory journey, ensuring your medical devices reach every corner of the globe, making a difference where it's needed most. Your vision is our mission, and together, we can shape a healthier future.
FAQs
Q1. Is it possible to sell medical devices internationally?

Yes, it is possible to sell medical devices internationally, but it requires compliance with the regulations of each target country. This may involve obtaining approvals, licenses, or certifications in multiple jurisdictions and ensuring compliance with local labeling and language requirements.
Q2. Are there international standards for medical devices?
Yes, various international organizations, such as the International Organization for Standardization (ISO) and the International Electrotechnical Commission (IEC), provide standards and guidelines for medical devices. Compliance with these standards is often required or recommended.
Q3. How long does it typically take to get approval or licensing for medical devices globally?
Approval or licensing time for medical devices can vary widely based on factors such as the country's regulatory framework, device complexity, and completeness of the application. It can range from several months to several years.